
Hace casi once años escribí una primera entrada sobre el chocolate, que casi nadie ha leído, centrada en lo que os voy a contar en esta, una necesaria actualización. La entrada nació de la lectura del interesante libro de Robert L. Wolke, "Lo que Einstein le contó a su cocinero", además de algunas búsquedas de trabajos científicos en torno a lo que se conoce como afloramiento (blooming en inglés) en la superficie de los productos a base de chocolate. Un problema complejo que ha dado muchos quebraderos de cabeza a la industria chocolatera. Se trata de ese fenómeno que es sobre todo evidente cuando el chocolate se ha conservado en unas circunstancias en las que ha sufrido continuos cambios de temperatura (como cuando ha estado en el interior de un vehículo, expuesto al sol de día y al frío de noche). Esa "agresión" genera una superficie blanquecina sobre nuestra tableta o bombón, además de otros efectos ligados a su textura y sabor, que siguen siendo estudiados hoy en día con las más modernas técnicas instrumentales, tanto por laboratorios universitarios como por los de las grandes empresas de alimentación.
Desde que escribí aquella entrada, se ha publicado mucha información adicional al respecto. Por ejemplo, hace pocos años me regalaron la segunda edición del libro de Stephen T. Beckett, "The Science of Chocolate", un texto muy interesante para los que, de alguna forma, hemos estado implicados en los complicados procesos de cristalización de materiales tan distintos del chocolate como son los polímeros o plásticos semicristalinos. Como es el caso de mi colega y amigo Alejandro J. Müller quien, con otros investigadores, publicó en 2013 un interesante artículo sobre asuntos relacionados con lo que nos ocupa [Journal of Food Engineering 116 (2013) 97]. De información como la citada se desprende que la cristalización y la reología del llamado licor de chocolate, que ahora explicaremos qué es, no tiene nada que envidiar en cuanto a complejidad a las de los polímeros. Y, concretamente, en lo que se refiere a la cristalización y a su evolución en el tiempo, cuando no se controla adecuadamente, es donde está el origen del temido blooming.
La fabricación del chocolate se inicia a partir de las almendras del cacao, contenidas en el interior de unas bayas leñosas o mazorcas, que nacen directamente de la planta tropical correspondiente. Tras su fermentación, secado y tostado (para incrementar aromas y colores mediante reacciones Maillard), la posterior molienda provoca que funda la grasa vegetal contenida en las almendras (conocida como manteca de cacao y que constituye el 55% de ellas) y se mezcle con partículas sólidas provenientes de la cubierta de las mismas y que se generan en la molienda. El resultado es una compleja mezcla con apariencia de un líquido espeso, oscuro y amargo, que es el licor de chocolate arriba mencionado. A partir de él son posibles varias alternativas. Una de las más comunes supone añadir a ese licor una importante cantidad de azúcar y cantidades variables de leche en polvo para obtener una nueva mezcla líquida que, adecuadamente enfriada, genera el clásico chocolate con leche. Si al obtener el licor de chocolate eliminamos las partículas sólidas provenientes del pulverizado de las almendras, que le dan su color marrón, entonces la posterior mezcla con azúcar y leche y su solidificación da lugar al chocolate blanco. Pero eso supone eliminar también los antioxidantes contenidos en las partículas sólidas, con lo que, en general, el chocolate blanco no se conserva tan bien como el chocolate más o menos oscuro y, como consecuencia, es mejor que no le de mucho la luz, que aceleraría la descomposición de las grasas de la manteca de cacao.
Sea cual sea la mezcla final, la cohesión entre los abundantes cristales de azúcar, que son hidrofílicos (se disuelven en agua), los sólidos derivados de las almendras y la manteca de cacao, que es hidrófoba (odia el agua), no es nada fácil. Por eso, los maestros chocolateros adicionan los llamados emulsificantes (el más habitual es la lecitina) para lograr la cohesión entre esas fases de distinto signo en el proceso conocido como conchado. En él, la mezcla de todos los componentes se calienta entre 55 y 75º y se mantiene en constante agitación para conseguir una emulsión estable (una especie de mayonesa a base de manteca de cacao). El proceso implica, al mismo tiempo, la eliminación progresiva de parte de la humedad contenida, así como de ácidos volátiles, probablemente el más importante de los cuales sea el ácido acético (vinagre).
Finalmente, la mezcla se deja enfriar de forma y manera que la manteca de cacao forme un sólido que cristaliza y cementa la mezcla con azúcar y partículas. Pero la cosa no es tan sencilla como enfriar agua para que se nos formen cristales de hielo a 0ºC y, por ello, necesita de un adecuado control en una fase de la fabricación conocida como templado. Y ello es así porque, a su vez, la manteca de cacao es una compleja mezcla de triglicéridos, moléculas a base de ácidos grasos y glicerina (como los de las grasas que, en el post anterior, daban lugar al jabón con el concurso de la sosa cáustica). Los ácidos de los que provienen esos triglicéridos pueden ser saturados (sin dobles enlaces) o insaturados (con dobles enlaces). Pues bien, la temperatura de cristalización de un triglicérido disminuye con el número de dobles enlaces. Adicionando leche a la mezcla, el contenido en insaturados crece y la temperatura de fusión disminuye. Por eso el chocolate negro, sin leche o con poca, necesita más temperatura para fundir que el chocolate con más leche.
Como consecuencia de esa complejidad, el enfriamiento de la mezcla de cacao, azúcar, las partículas sólidas procedentes de la molienda y, en su caso, leche, puede proporcionar un chocolate sólido que puede cristalizar hasta en seis formas cristalinas diferentes (lo que los químico-físicos llamamos polimorfismo de un sólido) y que se suelen denotar con números romanos entre el uno y el seis. Formas polimorfas son también, en el caso del carbono, los diamantes y el grafito de los lápices, ambos provenientes de un mismo magma fundido de carbono pero que se ha enfriado de forma diferente. Sólo una elección adecuada, por parte de los maestros chocolateros, de los procesos (templado) a los que se produce la cristalización, hacen que la mezcla fundida acabe cristalizando en la forma adecuada (la llamada forma V), cuyos cristales funden luego en nuestra boca a 37 ºC, unos pocos grados por encima de su temperatura de fusión a 33 ºC. La formación específica de esos cristales proporciona además esa apariencia satinada y atractiva de la superficie del chocolate, así como el agradable chasquido al partirlo.
Sin embargo, la estabilidad de esa forma V es muy complicada en las condiciones que, habitualmente, vive el chocolate en nuestras casas. Si el chocolate se mantiene a temperaturas un poco altas, la Forma V se transforma en poco tiempo en Forma IV, más blandita y que no produce chasquido al tratar de romperlo. Además, en esas condiciones, parte de la manteca de cacao puede estar ya en forma líquida y emigrar a la superficie de la tableta, donde "aflora" en forma de largos cristales que le dan la apariencia blanquecina del blooming. Si, por el contrario, el chocolate se mantiene a temperatura baja y mucho tiempo, por ejemplo en un frigorífico, la Forma V evoluciona a la Forma VI, un sólido más estable en esas condiciones. Es una transformación de un sólido en otro sólido, mucho más la lenta que la evolución de la Forma V a la IV. Pero, al final, la Forma VI también forma cristales en la superficie y el blooming aparece igualmente, aunque esta vez a mucho más largo plazo.
El afloramiento de cristales de triglicéridos es el fenómeno más habitual, aunque también puede ocurrir que algo del azúcar, existente en el chocolate en forma amorfa (no cristalina), emigre a la superficie y acabe cristalizando en ella con un efecto que, visualmente, es casi idéntico al afloramiento o blooming de la manteca de cacao. Hay una forma sencilla para distinguir si uno u otro proceso es el que ha dado lugar a la capa blanquecina que vemos. Basta con colocar una pequeña gota de agua sobre esa zona. Si el problema es debido a la manteca de cacao la gota se queda tal cual ya que no puede disolver a la manteca. Si es debido al azúcar, la gota se extiende sobre la superficie, a medida que el agua va disolviendo al azúcar.
Aunque los maestros chocolateros tratan de minimizar estos problemas con variados trucos en los que la Química Física de la fusión y cristalización de la manteca de cacao se maneja y controla adecuadamente, lo cierto es que mas vale que dejéis el chocolate en un sitio fresco, sin grandes variaciones de temperatura, y lo consumáis con una cierta rapidez (con peligro para vuestro peso). En caso contrario, el blooming os acecha.

Aunque, dadas las fechas, la foto de la izquierda pueda parecer una pila de diferentes turrones de mazapán, lo cierto es que son jabones. Con ocasión de dichas fechas hay casi un mercado navideño en cada ciudad (debajo de mi casa lo andan ya montando) y, en ellos, nunca faltan puestos de artesanos del jabón que nos venden todo tipo de versiones de jabones "naturales", "hechos en casa", "sin químicos" y otra serie de retahílas de las que, cada día, pueblan las informaciones sobre productos relacionados con la cosmética. Pero hay más cera que la que arde en esto de la Química y los jabones y, aunque ya he escrito sobre ello en otra ocasión, recientemente y como consecuencia de mi creciente interés por los perfumes, he tirado del hilo y he podido renovar mis opiniones al respecto.
La Food and Drug Administration (FDA) americana distingue claramente en una página al respecto entre los términos jabón y detergente. El jabón (el de siempre) lo define como la combinación de grasas (sólidas o semisólidas) y aceites (líquidos) (ya sean animales o vegetales) y un álcali, generalmente sosa o potasa cáusticas (NaOH y KOH para los químicos). Por el contrario, los detergentes son preparados también destinados a la limpieza pero obtenidos, en su gran mayoría, a partir de sustancias químicas de síntesis. A veces, en estos últimos, aparece la palabra jabón pero dice la FDA que no es un verdadero jabón de acuerdo con los términos regulatorios.
Pero, en esta definición, se deja claro que, para que haya jabón, tiene que haber un álcali. Si uno quiere hacer un jabón en pastilla generalmente se usa la sosa, si lo quieres más blandito la potasa. Pero si no hay un álcali en juego no hay jabón. Eso lo reconocen hasta las páginas webs más radicales sobre la vida "natural". Así que, punto uno, un jabón no existe si no hay una reacción química (llamada saponificación) entre grasas o aceites con álcalis. En cuanto a las grasas y aceites, ahora lo que privan son los de origen vegetal: el aceite de oliva, el aceite de coco, el aceite de girasol, al aceite de palma (este hoy un poco desprestigiado), el aceite de canola y otros muchos más provenientes de plantas exóticas (como el de aguacate). Pero, no hay que olvidar que una gran parte de los jabones convencionales que se han hecho en este país lo han sido a partir de grasas animales como el sebo, un subproducto del sacrificio de vacas y cerdos.
Tras la adecuada reacción química con los álcalis esas grasas y aceites se transforman en nuevos productos que aparecen listados en los ingredientes de los jabones y que, curiosamente, incluso en los que que se venden en España, lo hacen con sus nombres en inglés. Por ejemplo, en el famoso y conocido jabón de Heno de Pravia, la cara anterior del envase está redactada en castellano y contiene los términos Original y Jabón Natural. Pero en la parte trasera (por lo menos en el que yo he comprado en mi súper) aparecen los ingredientes del mismo en inglés: Sodium tallowate, sodium cocoate, water, glycerin y otra larga serie de ingredientes correspondientes a aromas, conservantes, etc. Sodium tallowate es el resultado de la reacción del sebo de vacuno con sosa cáustica, de donde proviene el sodio. Sodium cocoate es el resultado de la reacción del aceite de coco con sosa cáustica. En otros jabones se pueden leer términos como sodium olivate, resultado de la acción de la sosa cáustica sobre el aceite de oliva. Como las grasas y aceites son mezclas bastante complejas de moléculas químicas conocidas como triglicéridos, se puede concluir que, punto dos, los componentes esenciales del jabón, tal y como finalmente se venden, son nuevas y complejas mezclas de sustancias químicas derivadas de las reacciones con los álcalis de las grasas y aceites empleados, aunque os lo quieran vender como si estuviera compuesto por las propias grasas o aceites extraídos de plantas naturales.
Otro punto interesante es el asunto de si algo tan corrosivo como la sosa cáustica sigue perviviendo en el jabón tras el proceso que ha dado lugar al mismo. Las páginas web que ilustran sobre cómo fabricar jabón suelen contener calculadores para estimar la cantidad exacta de sosa cáustica que hay que emplear para estar seguros de que toda esa sosa se consume, dependiendo del tipo de grasa o aceite que vayamos a emplear para fabricar el jabón. Y si se consumiera por completo, sería lógico que no apareciera en la lista de ingredientes como tal, ya no está en el jabón. Pero esos cálculos solo son aproximaciones groseras, ya que dentro de un mismo tipo de grasa o aceite (pongamos el caso del aceite de oliva), la composición puede variar dependiendo del origen, tipo de aceituna, procesado, etc. y es muy probable que el ajuste que proporcionan esos calculadores haga que, en el jabón final, quede algo de aceite o de sosa cáustica sin reaccionar. No es nada peligroso porque, en general, son cantidades pequeñas que incluso pueden ir desapareciendo por la acción del CO2 del ambiente. Pero si el exceso de sosa cáustica es importante podría causar problemas a las pieles sensible. De hecho, hay normas sobre la cantidad máxima de sosa cáustica que puede contener un jabón comercial y procedimientos analíticos para determinarla. Así que, punto tres, el jabón de toda la vida, si no está bien hecho, puede contener restos de sosa cáustica y pudiera causar problemas (leves) de los casi ningún fabricante quiere hablar. Unos pocos, más honestos, reconocen que puede quedar sosa en sus jabones y en las etiquetas aparece como ingrediente su nombre químico, hidróxido sódico o sodium hydroxide.
Y, finalmente, hay gente que en estos foros de cosmética que ando visitando hacen preguntas de lo más lógicas. ¿Cómo se puede adjetivar a estos jabones como "naturales" cuando usamos un "químico" tan enérgico como la sosa cáustica y obtenemos nuevos productos que no están generalmente en la naturaleza?. Buena pregunta, pardiez. Aquí los fabricantes usan todo tipo de estratagemas. La más común es decir que la sosa desaparece durante la producción del jabón algo que, al menos en parte, acabamos de desmontar. Pero, aunque así fuera, eso mismo le pasa al Bisfenol A al producir policarbonato o resinas epoxi y ya véis la que hay montada sobre él. Hay quien argumenta que la sosa cáustica no puede considerarse "no natural" porque se obtiene, fundamentalmente, a partir de la electrolisis de disoluciones de sal común (cloruro sódico), algo que está en la naturaleza, sin emplear disolventes ni otros reactivos de carácter sintético. Respuesta que tiene su mérito, pero sin ningún rigor químico. En cualquier caso, todos ellos obvian que los sebatos de sodio, los olivatos de sodio o los cocoatos de sodio de las etiquetas de ingredientes (ahora los he puesto en castellano) son sustancias de síntesis que aparecen como consecuencia de la reacción de las grasas o aceites con la sosa cáustica. Y el que no lo quiera ver así es porque tiene intereses en ello.
Y ya mejor no os empiezo a contar cosas de jabones para veganos, jabones sin gluten o jabones hechos en casa con restos de aceite usado para freír, porque esto me quedaría muy largo y luego me riñen.
P.D. El que los ingredientes aparezcan en inglés no es tan curioso. Es un problema de normativa europea, como muy bien explica el interesante comentario que, muy de mañana, me ha colocado el Prof. Mans y que aparece debajo.

Pues eso. Que esta es la quingentésima entrada de este Blog, de acuerdo con las cuentas que echa el editor Blogger. No pensaba escribir nada que denotara tamaña efeméride pero, en las últimas semanas, ha habido una cierta movida en Twitter entre conocidos blogueros a los que llevo siguiendo desde hace tiempo y que se han ido planteando diversas preguntas en torno a este tipo de actividad. Así que me ha parecido procedente contar mi propia visión de algunos de los temas que han ido saliendo. Como, por ejemplo, ¿por qué sigo escribiendo este puñetero Blog?.
Vaya por delante que pienso que, en el ámbito de la actividad bloguera dedicada a la divulgación científica, hemos sobrepasado ya un cierto máximo de apogeo y vamos camino de una autorregulación natural a la baja, consecuencia de los muchos cambios que se han producido a lo largo de los últimos años en este ámbito. La divulgación científica se ha profesionalizado (dentro de límites modestos) y los blogs más afamados, aquellos que han sabido cautivar y seguirán cautivando a la gente mientras el autor tenga ganas para seguir escribiendo, son, en muchos casos, escaparates de la actividad casi profesional de sus autores, algo que tiene poco que ver con lo que otros muchos hacemos.
Yo empecé con las entradas de este blog un 28 de febrero de 2006. Estaba en un momento delicado de mi vida académica. Los años empezaban a pesar y los cambios cada vez más acelerados en la manera de hacer Ciencia me estaban haciendo mucha pupa. Por otro lado, mis largos años de docencia me habían dado mucha materia para enganchar a mis estudiantes en la repercusión de la Química en nuestra vida diaria y para desmontarles muchos de los bulos que la Quimiofobia rampante iba introduciendo en sus jóvenes e influenciables mentes. Y luego estaba el gusanillo de escribir, latente desde que dejé de mandar casi una misiva diaria a la Búha en mis tiempos de estudiante en Zaragoza y, posteriormente, en largos intercambios epistolares con un amigo del alma. La aparición del email anestesió esta pasión por escribir durante un tiempo, porque la facilidad que da el escribir y enviar un email siempre deja espacio para toques personales más o menos "literarios", por muy profesional que sea el contenido de los mismos. En otras palabras, yo siempre me he enrollado en mis emails.
Pero llegó un momento que tocaba buscarse una nueva razón para escribir con más fundamento y opté por lo que los más viejos seguidores de este Blog han conocido a lo largo de los años. Así que, esencialmente, escribo porque me gusta escribir (lo haga bien o mal), porque ello me permite (y más ahora, como jubilado) tirar del hilo de temas que me preocupan o interesan y estudiarlos, sin prisas, para hacerme con una opinión al respecto, al margen de lo que sea política o científicamente correcto. Eso me ha dado una carrocería mental, yo diría que bastante escéptica, sobre la mayor parte de los temas con los que me encuentro en el día a día. Y, lo que ahora es muy importante, me hace casi trabajar a media jornada (para la otra media tengo otras devociones).
De alguna manera, por tanto, comparto la opinión de Francis Villatoro, que dice que escribe en su Blog para aprender sobre los temas que le interesan. Y con ello no me opongo a la versión de mi amigo José Manuel López Nicolás, que le dio un giro semántico a la frase de Francis diciendo que él aprende divulgando. Muy sutil, pero eso no define por qué escribe divulgación sino qué le ocurre mientras lo hace. Conociendo a Jose sé que lo hace porque, desde el principio (y me conozco sus principios), su desbordante personalidad mediterránea disfruta divulgando, sea escribiendo el blog o impartiendo conferencias.
Lo que si sé es que yo nunca me he planteado esto como un oficio, entre otras cosas porque, como oficio, no tiene "salida", al menos en los parámetros en los que yo me lo planteo. Me da una pereza inmensa, por ejemplo, tratar de poner parte de mis entradas en forma de libro (como el amigo José Miguel Mulet y otros han hecho de forma tan exitosa). Ni quiero tampoco convertirme en un itinerante a lo largo y ancho de la piel de toro, compartiendo mis disquisiciones con los forofos de la Ciencia. No me importa colaborar y dar alguna charla ocasional donde me llamen pero, a ser posible, lo suficientemente cerca de mi casa como para dormir en mi cama.
También he tenido siempre claro que mis entradas no eran para especialistas en Química. Evidentemente, uno tiene sus sesgos y, más de una vez, se me ha ido la mano y he tenido que sufrir los rapapolvos de mis lectores más próximos, con comentarios del tipo "te has pasao, muy técnico...". Pero mi intención ha sido siempre la contraria. Algunos en las RRSS han bautizado ese punto de vista como "escribir para que te entienda tu abuela". En mi caso, vamos a dejarlo en escribir para que me entienda "la gente de mi edad" que para eso tengo ya una edad provecta. No me parece mal que haya Blogs más técnicos, dedicados a temas muy especializados (¡faltaría plus!), pero ese no es el nivel al que yo quiera (y pueda) divulgar. Aunque rebajar mucho el nivel de un tema no siempre es tarea fácil y, a veces, tiene el peligro de que tus pretensiones se acaben convirtiendo en una caricatura. Mi admirado Pedro M. Etxenike, cuando hemos hablado de los problemas de divulgar, me ha mencionado más de una vez una frase de Einstein “Todo debe hacerse tan sencillo como sea posible. Pero no más”. Aunque el mismo Einstein también parece que decía que "Si no lo puedes explicar de forma sencilla, es que no lo has entendido bien". Y por ahí creo que anda el reto de cada cual.
Lo que si tengo claro es que no llegaré a escribir otras quinientas entradas en lo que me queda de resuello.

A mi con lo del perfume me pasa como con el vino. A ambos objetos de placer los descubrí gracias a mi familia política y, en años posteriores, mis conocimientos al respecto se agrandaron gracias a ellos. Tanto de uno como del otro guardo buenos recuerdos de mis primeras experiencias. Y, en el caso de los perfumes, aunque mi memoria empieza con el agua de colonia que usaba mi madre con nosotros y la primera fragancia que usaba la Búha en nuestro primeros tiempos juntos (Agua Brava), lo cierto es que mi recuerdo inicial más largo es el uso que ella hacía de un perfume de Dior, comercializado en 1966 bajo el nombre de Eau Savage. Lo que demuestra que mi chica ha sido siempre un poco rarita, porque Dior creó esa fragancia para el nuevo tipo de hombre que parecía emerger en los convulsos sesenta y que, de alguna manera, resume el genial cartel publicitario debido a René Gruau, que veis a la izquierda.
El desarrollo del perfume fue encargado por Dior al famoso perfumista Edmond Roudnitska, un auténtico nariz de oro, quien mezcló toques de cosas clásicas como el limón, el romero, la lavanda, la albahaca, el vetiver y otros, pero imprimió su genialidad en el nuevo perfume adicionando una cantidad importante de un componente nunca utilizado en perfumería (aunque sobre esto hay alguna duda), una sustancia de síntesis conocida por los químicos como metil dihidro jasmonato, a la que se bautizó con el nombre comercial de Hedione. Esa molécula añadía una potente sugerencia de jazmín y, sobre ella, voy a componer esta entrada porque creo que puede resultar interesante no sólo para los químicos sino también para los interesados en los perfumes.
Las flores y las plantas siempre han constituido el componente fundamental de la perfumería, aunque la llegada de las fragancias sintéticas ha ido cambiando esa percepción. Y ello ha sido así por razones económicas pero también porque la Química ha permitido identificar y reproducir los aromas atractivos de determinados productos de la Naturaleza. Y voy a poner como ejemplo, al hilo de la Eau Sauvage, el jazmín mediterráneo (Jasminum grandiflorum). Considerado uno de los aromas más populares ("ningún perfume sin jazmín" ha sido una frase típica en este ámbito), el problema es que las flores de jazmín se recolectan a mano (y tempranito por la mañana) y hace falta la friolera de una tonelada de estas flores para producir algo más de dos kilos de lo que se conoce como concreto de esa fragancia, una pasta obtenida tras poner las flores en maceración en un disolvente o mezcla de disolventes (para que las ceras y esencias de la planta se diluyan) y evaporar posteriormente el o los disolventes. Después, este concreto se disuelve en alcohol y se destila para conseguir el absoluto, un líquido que es la esencia "pura" de la planta en cuestión. En el caso del jazmín, la tonelada inicial no da finalmente casi ni para un kilo de absoluto asi que, en los años 50, la producción mundial de absoluto de jazmín no llegaba a las seis toneladas lo que, dada la demanda del producto, ponía el precio del mismo por las nubes.
Para esa misma época, el 87 por ciento de los componentes de ese aceite esencial de jazmín estaba ya identificado pero los químicos parecían no dar con la molécula responsable del carácter profundamente floral de este absoluto. Así que la firma Firmenich, una empresa suiza líder en el mercado de la perfumería, decidió financiar una Tesis en el Instituto de Biologie Physico-Chimique de Paris para que un joven Edouard Demole investigara en profundidad el concreto del jazmín, a la búsqueda de alguna evasiva molécula que constituyera el singular carácter de las notas de jazmín. En 1957 Demole consiguió su objetivo, aislando una molécula denominada metil jasmonato, que fue correctamente identificada sobre la base de las técnicas instrumentales existentes en la época. Pero en esos procesos de identificación del metil jasmonato, Demole llevó a cabo una reacción sencilla de ese compuesto con hidrógeno, obteniendo el antes mencionado metil dihidro jasmonato. Este producto huele también a jazmín aunque menos que el jasmonato sin hidrogenar, pero tiene ciertos tonos cítricos que su pariente no tiene. Y sobre todo, Firmenich comprobó por otras vías que su síntesis era mucho más fácil y barata que la del jasmonato de las flores de jazmín. Ambas moléculas fueron sintetizadas a finales de los cincuenta y patentadas en 1960, tras lo que la empresa suiza empezó a mandar muestras a perfumistas conocidos, entre ellos Roudnitska, que enseguida utilizó el Hedione o jasmonato hidrogenado en mi recordada Eau Sauvage, empleando una concentración bastante grande para lo que se emplean los componentes en perfumería (un 2,5%).
Hoy en día, la producción de Hedione se ha abaratado considerablemente y ha dejado de ser un componente exclusivo de perfumes más o menos caros como Chanel Nº 19, First de Van Cleef o Cristalle de la misma Chanel. Pero la investigación en torno a esa molécula no ha cesado y se han desarrollado otras moléculas relacionadas, como el Hedione C, una variante mucho más rica en el isómero cis (75%) que la del Hedione a secas, que solo contiene un 10% de ese isómero.
Pero esto ya son cosas para el consumo de la chusmarra de químicos a la que he pertenecido y que hoy celebramos nuestra fiesta de San Alberto. Particularmente para los que se dedican a la síntesis orgánica. Aquí les dejo un link por si alguno de ellos quiere ilustrarse más en el tema.

En el año 2004 la entonces Ministra de Medio Ambiente, Cristina Narbona, se adhirió a la campaña DETOX de la organización ecologista WWF/Adena (hoy WWF España) haciendo analizar su propia sangre y la de los altos cargos de su Departamento, a la búsqueda de una larga serie de sustancias químicas, consideradas tóxicas y, que según manifestó entonces la Ministra "son la cara oculta del progreso". No esconderé que no me hizo ni pizca de gracia esa adhesión, porque las noticias se fijaron en que todos los analizados tenían en su sangre una media de treinta y tantos de esos productos químicos pero, por mucho que busqué, en ningún sitio pude encontrar las cantidades detectadas, algo fundamental para saber si la Ministra estaba realmente intoxicada o no. Pero bueno, a lo que iba que me lío, en la lista de sustancias a buscar en los cuerpos de nuestr@s polític@s tenían un peso fundamental una serie de sustancias (y familias de sustancias) cuya característica fundamental es tener muchos átomos de cloro y/o bromo en su estructura. Ese es el caso del mismísimo DDT, las dioxinas y sus primos los furanos y los también famosos policlorobifenilos (PCBs para los amigos) y polibromo difenil éteres (PBDEs), estos dos últimos usados en diferentes aplicaciones contra el fuego. Todos los citados han sido prohibidos desde hace bastantes años pero siguen estando en candelero porque, además de ser tóxicos, comparten otra característica que les hace peligrosos y es que se acumulan, y de forma persistente, en el organismo de los seres vivos. Es lo que se conoce como Contaminantes Orgánicos Persistentes (o por sus siglas inglesas POPs).
En el año 2011, un artículo del Chemical and Engineering News (CEN) firmado por Carmen Drahl llamó mi atención. Se resumía en él un trabajo publicado por científicos del Servicio de Salud y Seguridad Alimentaria del Estado alemán de Baviera, en el que daban cuenta del hallazgo fortuito de niveles relativamente altos de un antibiótico natural, la Drosofilina A, en la carne de jabalíes salvajes. Esa molécula es producida por un tipo de hongo conocido como Agaricus subatratus que parece gusta mucho a los amigos de Obelix. De nombre químico 2,3,5,6-tetracloro-4-metoxifenol, tiene la característica de poseer cuatro cloros en su molécula y parece acumularse en el cuerpo de los cochinos, porque las concentraciones medidas por los bávaros eran mucho más altas que las correspondientes al DDT, igualmente analizado por el equipo. No tiré mucho más del hilito en esa ocasión porque del artículo parecía desprenderse que la sustancia no era nociva para los jabalíes. Además, terminaba con una de esas frases con las que casi siempre acaban los artículos científicos, diciendo que comprender el mecanismo por el que la Drosofilina A no parece afectar a los jabalíes sería un buen punto de partida para el diseño de compuestos halogenados (como llamamos los químicos a los que tienen cloro y bromo) menos perjudiciales para el medio ambiente. Siempre me quedó la duda de la cantidad de Drosofilina que aparecería en un hipotético análisis de la sangre de Obelix.
Pero mañana lunes, el citado Chemical Engineering News (CEN) publica otro artículo (que ya lleva unos días en su versión web) que tiene que ver con compuestos halogenados de origen natural y que me ha permitido conocer una serie de hechos que no me resisto a contaros. El artículo está firmado por Deirdre Lockwood, quien comienza la historia contando cómo, a finales de los noventa, se encontró en huevos de una serie de aves marinas una molécula que, resumidamente, podríamos llamar un bipirrol halogenado, cuya estructura recuerda la de los PCBs ya entonces prohibidos y muy buscados por los investigadores en la vida animal y en el medio ambiente. Como la molécula no había sido registrada ni se encontraba en la literatura, la conclusión era que tenía que haberse originado en la Naturaleza, afirmación que los investigadores confirmaron mediante estudios de carbono-14, un isótopo radiactivo del carbono normal, que se va descomponiendo a lo largo de los años. La relación entre ese carbono-14 y el carbono total permite datar la fecha de síntesis de un determinado compuesto. Y así, si nuestro bipirrol hubiera sido generado a partir de sustancias provenientes del petróleo, formado hace miles de años, esa relación tiene que ser sustancialmente diferente de la de un bipirrol generado hace mucho menos tiempo por algún organismo natural.
Pero este bipirrol halogenado no es una rara avis en la Naturaleza. Compuestos polihalogenados (con varios cloros y/o bromos) como la Drosofilina A no son muy corrientes en el medio terrestre pero si en el mar. El artículo cita un libro en el que se recogen hasta 6000 compuestos de ese tipo que, fundamentalmente, se han encontrado en el mar. Uno de ellos es otro bipirrol con siete átomos de cloro (denotado como Q1) que se ha encontrado en la leche materna de varias mujeres de las Islas Faroe, en cuya alimentación hay un componente importante, la grasa de ballena, en la que el Q1 se acumula. Todavía no hay estudios de toxicidad concluyentes sobre estos bipirroles pero los investigadores sospechan que debe ser similar a sus "primos" sintéticos a los que se parecen.
Otro ejemplo de compuesto, este con cuatro bromos y denotado como 6-OH-BDE-47, es muy parecido a los PBDEs que se introdujeron en los setenta como retardantes a la llama, sustitutos de los entonces recién prohibidos PCBs y que, finalmente, también acabaron prohibidos. Este compuesto aparece en un tipo de esponja marina como la que veis en la foto que ilustra esta entrada y que pertenece a la familia de las Dysideidae. La presencia de esta sustancia en la esponja es importante y llega alcanzar el 10% de la misma, una vez seca. Estudios posteriores, sin embargo, demostraron que no es la propia esponja la que sintetiza el compuesto, sino una especie de bacteria simbiótica que vive en la esponja (Hormoscilla spongeliae) y que lo usa para protegerse de sus depredadores. De nuevo, tampoco se tienen por el momento datos concluyentes sobre su toxicidad pero, al igual que con los bipirroles, si sabemos que se acumula en el cuerpo humano, igual que sus parientes PBDEs, y que ya ha sido también detectada en la sangre de embarazadas coreanas y en el cordón umbilical de sus fetos en concentraciones del orden de las partes por trillón.
El asunto como veis es interesante y deja varios interrogantes en el aire, desde las cantidades de sustancias cloradas que la Naturaleza pone en el medio ambiente, a la posible toxicidad de muchas de ellas, que nadie ha estudiado porque hemos andado atareados en las producidas por el hombre. También permite abordar el problema de por qué algunos microorganismos son capaces de destruir eficientemente sustancias como los PCBs. Como dice un investigador citado en el artículo del CEN, llevan muy pocos años en esa labor de limpieza y no parece que sus genes hayan tenido tiempo todavía para adaptarse a esa labor. Lo que implicaría que se trata de una capacidad que han adquirido a lo largo de los tiempos y que sería interesante investigar para sacar conclusiones y nuevas estrategias para eliminar esas sustancias tan resistentes a desaparecer del medio ambiente. Y, finalmente, queda muy evidente la magnitud del “arsenal químico” que nos queda por descubrir en el fondo del mar. Para bien y para mal.
A mi el artículo me ha encantado y voy a utilizarlo en una charla del próximo 6 de noviembre para mostrar la difusa frontera que separa lo natural de lo sintético.

La controversia entre los partidarios y detractores de la homeopatía, que ya dura más de un siglo, tiene un antes y un después de la introducción de la llamada "memoria del agua", tras la publicación en Nature, el 30 de junio de 1988, de un artículo firmado por el grupo liderado por el médico e investigador francés Jacques Benveniste. Dicho artículo trataba de la activación de ciertos glóbulos blancos (daré más detalles después) por unas disoluciones de un anticuerpo que, como los mismos autores reconocían en el texto del artículo, eran tan diluidas que estaban más allá del límite de Avogadro o, contado para que todo el mundo lo entienda, se había diluido tanto la disolución de partida del anticuerpo que era imposible que esas altas diluciones contuvieran ni una sola molécula del mismo. Algo que tiene mucho en común con los remedios homeopáticos, una gran parte de ellos preparados a esas altas diluciones, antes de ser evaporados sobre los gránulos habituales.
Desde entonces, hace ya casi treinta años, médicos y científicos partidarios de la homeopatía siguen buscando el fundamento de esa posible "memoria" y, en años más recientes, la cosa se está poniendo casi imposible de seguir a quien no tenga unos conocimientos más que básicos de la estructura del agua líquida, de alta biología o, incluso, de mecánica cuántica. No llegaré a tanto en esta entrada. Al hilo de un par de libros que he leído recientemente en torno a Benveniste y la "memoria del agua", me propongo contaros la historia previa a la publicación del famoso, y polémico, artículo que dio lugar a ese concepto. Tiempo habrá para considerar las posteriores derivas del asunto.
A principios de este año Guillermo Peris, un Profesor de la Universitat Jaume I, al que sigo en Twitter, me comentó en una conversación privada que estaba leyendo un libro aparecido en 2016 bajo el título "Ghosts of molecules", escrito por Francis Beauvais, uno de los firmantes del mencionado artículo de Nature. Se trata de la versión en inglés de la segunda edición francesa, aparecida en 2015, que corregía y aumentaba la primera edición, publicada también en francés en 2007. Se puede descargar gratuitamente aquí esa segunda edición en ambos idiomas. El libro está estructurado en dos partes bien diferenciadas que tienen como frontera el acontecimiento de la propia publicación del artículo de Nature. Lo he leído y releído varias veces desde entonces y, gracias a él, he descubierto otro libro, una obra póstuma del propio Benveniste, publicado por tres de sus hijos bajo el título "Ma verité sur la memoire de l'eau" que, en formato pdf y en francés, os podéis descargar gratuitamente aquí.
A través de ellos he descubierto la compleja personalidad del citado Jacques Benveniste, alguien que quería ser piloto de carreras en su juventud y estudiar Ingeniería Mecánica para estar cerca de los coches pero que, finalmente y por una serie de razones, acabó estudiando Medicina. Tras un periodo de diez años como médico en diferentes Hospitales de París, comenzó a simultanear su trabajo de médico con el de investigador en Inmunología en el Instituto del Cáncer (CNRS) de Villejuif. En esa situación le pilló la atmósfera previa y posterior del mayo del 68 en el que, como militante del PS francés, tuvo sus más y sus menos con relevantes figuras del panorama médico e investigador del país de los galos, incluido el Premio Nobel de Medicina de 1965 André Lwoff. Otro hecho que muestra su carácter inconformista son sus críticas reiteradas (por ejemplo, en la Tribune Libre del periódico Le Monde) sobre el sistema de mandarines que controlaba el panorama médico y científico de la Francia del momento. Sea por ese ambiente irrespirable para él o por otras razones que no quedan claras, a principios de 1969 aceptó un contrato de la Scripps Clinic and Research Foundation en La Jolla, California, Institución dirigida por el Prof. Frank Dixon, uno de los pioneros de la inmunología.
A partir de ese momento inició lo que debería haber sido una exitosa carrera científica (y en parte lo fue), que puso sus cimientos en una serie de artículos en prestigiosas revistas sobre el llamado Factor Activador de Plaquetas (PAF), una especie de mediador entre los glóbulos blancos y las plaquetas, con importantes repercusiones en fenómenos ligados a las alergias y a otras patologías inflamatorias.
De vuelta en Paris, tres años después, con una aureola de investigador de prestigio pero controvertido en los medios de su país, Benveniste acabo creando una unidad de investigacion sobre "Inmunopatología de la alergia y la inflamación", la Unidad 200 del INSERM (Instituto Nacional de la Salud e Investigación Médica), que pronto creció en dinero y personal, y entre cuyas líneas estaba el estudio de los basófilos, un tipo de glóbulos blancos que juegan un papel importante en las reacciones alérgicas. En presencia de alérgenos como el polvo, el polen, la clara de huevo, etc., esos basófilos pierden unos gránulos existentes en su citoplasma (un proceso llamado degranulación), liberando al mismo tiempo ciertas sustancias como la histamina.
En esa unidad apareció a principios del año 1980 Bernard Poitevin, que finalmente también firmaría el trabajo de Nature, un estudiante de doctorado que había empezado su Tesis sobre el mediador PAF descubierto por Benveniste. Pero Poitevin era también médico homeópata y entre finales del 81 e inicios del 82 propuso a Benveniste realizar experiencias sobre los efectos de los preparados homeopáticos en modelos biológicos de laboratorio. Benveniste le dejó hacer, entre otras cosas porque Poitevin venía con dinero debajo del brazo, de la mano de los Laboratorios Homeopáticos de Francia (LHF). En esas mismas fechas, Boiron, una de las grandes empresas de homeopatía hoy en día, se puso en contacto con Benveniste para tratar de reproducir en su laboratorio ciertos resultados obtenidos por Jean Saint-Laudy (que también aparece como autor en el artículo de Nature), un médico de un laboratorio privado que había utilizado un método ideado por Benveniste para contar, bajo un microscopio, los basófilos que se degranulaban. Boiron acabó firmando otro contrato con Benveniste en 1983, año en el que esa empresa absorbió a los Laboratorios Homeopáticos de Francia. Hay que decir también que, en esa época, el Laboratorio de Benveniste recibía la parte más relevante de su financiación exterior de importantes empresas farmacéuticas.
Así que ya tenemos un pequeño germen de experiencias homeopáticas en el seno del Grupo de Benveniste. Saint-Laudy trabajando sobre el efecto inhibidor de la histamina a altas diluciones en la degranulación de basófilos y Poitevin en el mismo efecto pero utilizando para ello un conocido preparado homeopático, Apis Mellifica, obtenido a partir de un extracto (o tintura madre) en alcohol de abejas machacadas y diluido muchas veces. Según las memorias de Benveniste, él no creía en absoluto en que una alta dilución de algo pudiera tener actividad biológica pero cuando Poitevin empezó a presentarle ciertos resultados que contradecían su opinión, empezó a picarle la curiosidad. En setiembre de 1984 Pointevin presentó sus primeros resultados sobre la Apis Mellifica en un Forum de Jóvenes Investigadores y, en enero de 1985, Benveniste impacta a los medios franceses cuando, en una Mesa Redonda organizada por la revista Impact-Medicine, dice que las altas diluciones ya no son un problema de la homeopatía y, para avalar esa opinión, presenta los resultados de Pointevin. Todo hay que decirlo, la Ministra francesa de Sanidad de la época era favorable a las medicinas alternativas y a que los remedios homeopáticos se reembolsaran por la Seguridad Social francesa.
A principios de 1986, Benveniste hace circular, a nivel interno de su Grupo, un primer manuscrito que pretendía enviar a Nature y en el que se resumían los resultados de la inhibición de la degranulación de basófilos causada por la Apis Mellifica y la histamina a altas diluciones. En esa consulta interna, alguien apuntó los problemas que podría causar en el Consejo Editorial de Nature la mención en el artículo a preparados homeopáticos. Tomando en cuenta esa sugerencia, Benveniste decidió eliminar los resultados de la Apis Mellifica (que se enviaron a otra revista) y centrarse solo en la histamina a altas diluciones. Tras esos cambios, en junio de 1986, el artículo se envía a Nature, que lo rechaza en noviembre de 1986 pero abriendo la puerta a reconsiderarlo si otros laboratorios confirman los resultados.
A partir de ahí, en unos frenéticos 1987 y 1988, Benveniste y los editores de Nature mantienen una dura pelea que configura la versión final del artículo que finalmente se publicará en la revista. Por un lado, se realizan ensayos en otros laboratorios situados en Milán, Toronto y Tel-Aviv, con resultados no siempre favorables a las tesis de Benveniste, como ocurrió en Israel. Al mismo tiempo, se produce un cambio importante de estrategia que afectaría sustancialmente al contenido del artículo finalmente publicado. Durante las experiencias con la histamina, se adicionaba a las muestras de basófilos cantidades de un anticuerpo, el denominado anti-inmunoglobulina E (anti-IgE) para provocar una degranulación previa y poder ver así el efecto de las diluciones de histamina a la hora de inhibirla. En esos meses, el grupo de Benveniste cree tener resultados que muestran que la anti-IgE es capaz de activar la degranulación no solo a concentraciones habituales sino a muy altas diluciones, de nuevo por encima del límite de Avogadro. Eso hace que el interés del grupo pase de considerar altas diluciones de histamina que inhiben la degranulación a altas diluciones de anti-IgE que la provocan.
En agosto de 1987 Benveniste envía un segundo manuscrito que solo contenía resultados de la activación de la degranulación por altas diluciones de anti-IgE. El editor de Nature, John Maddox, contesta en noviembre rechazando el artículo. Benveniste se cabrea y lo manifiesta por correo y por teléfono y, de nuevo, Nature abre una puerta (finales de enero de 1988) pidiendo que se complete el artículo con una explicación de los mecanismos por los que una dilución tan alta, en la que no puede haber ni una sola molécula de anti-IgE, puede causar ese efecto de activación. Benveniste le contesta el 2 de febrero de 1988 en una carta en la que, como posibles causas, ya aparecen términos como "moléculas fantasmas" (que da lugar al título del libro que ilustra esta entrada) que "pudieran estar compuestas por moléculas de agua ordenadas previamente en torno a la molécula de anti-IgE".
Entre marzo y abril de 1988 hay nueva correspondencia y llamadas de teléfono entre Benveniste y Maddox y se introducen pequeños cambios en el texto. A finales de abril, Benveniste se va a las Bermudas a un Congreso en el que participan varios Premios Nobel, donde pronuncia una conferencia sobre las altas diluciones que, según él, atrajo el interés de muchos de ellos. En una nueva carta enviada desde allí a Nature, aparece una referencia al Prof. Emilio Dei Giudice, de la Universidad de Milán, autor de una teoría sobre "la organización de dipolos de agua, creando una campo electromagnético que puede reproducir el previamente originado por la molécula que estamos diluyendo".
El 27 de mayo de 1988 se celebra un Congreso Nacional de Homeopatía en Estrasburgo. Las crónicas enviadas desde allí por los corresponsales de Le Monde y Liberation parecen estar en el origen del término "memoria del agua". Le Monde reproduce frases de una rueda de prensa de Benveniste hablando de "un efecto molecular sin molécula" o de "moléculas fantasmas, de improntas moleculares, de un agua que habría conservado el recuerdo de las sustancias con las que ha estado en contacto”. La idea de que el agua pudiera tener memoria empieza a materializarse pero parece que el término exacto "memoria del agua" lo introdujo el corresponsal de Liberation en una crónica fechada el 30 de mayo. La personalidad desbordante de Benveniste fue más allá en esas declaraciones y llega a decir esos días que “El procedimiento que hemos usado es similar a agitar las llaves de un coche en el Sena, en el Pont-Neuf, y luego coger unas gotas del río en la desembocadura de El Havre y hacer que arranque ese coche y no otro”.
El caso es que, probablemente, el eco de las últimas declaraciones de Benveniste en los medios franceses fuera la causa por la que Maddox, el editor de Nature, diera su brazo a torcer y publicara el artículo el 30 de junio de 1988. En él, las altisonantes frases anteriores se matizan bastante y se resumen en una corta explicación sobre el sorprendente efecto de las altas diluciones: "El agua puede actuar como un “molde” para la molécula, por ejemplo a través de una red de enlaces de hidrógeno o merced a campos eléctricos y magnéticos. En el momento presente, sólo podemos especular sobre la naturaleza de la actividad específica presente en las disoluciones altamente diluidas".
A partir de la publicación del artículo empezaron las desgracias para Benveniste y su Grupo. El INSERM no le respalda en lo relativo a las altas diluciones y él tiene que admitir que una curiosa Comisión nombrada por Nature (con el mago Randi incluido) visite su laboratorio y asista a una repetición de los experimentos, que los comisionados consideraron llenos de errores experimentales y estadísticos. Y tras esos dos reveses y como él mismo cuenta: "A partir del otoño de 1988 y, en pocas semanas, me convierto en un paria de la Ciencia. Prácticamente ningún científico francés acepta ver su nombre junto al mío".... "En los medios de la homeopatía, no se me proporciona más que un soporte mínimo, por no decir altamente diluido. En junio de 1988 tras la euforia de la aparición del artículo, Boiron, cuyo Director científico es uno de los firmantes (Poitevin) del mismo, me había anunciado créditos ilimitados a mi disposición. Un mes más tarde, la Dirección de la empresa estima que si el INSERM no apoya mis investigaciones, no puede seguir financiándome. El dinero se corta brutalmente en junio de 1989. Las ratas abandonan el barco".
Benveniste no dio su brazo a torcer y siguió hasta su muerte defendiendo la validez de sus experimentos con las altas diluciones de moléculas de actividad biológica. Pero eso forma parte ya de la segunda parte del libro de Francis Beauvais y se queda para otra entrada que esta ya es larga y un poco prolija.

Raro es el día que los medios de comunicación no nos bombardean con alguna noticia relacionada con el calentamiento global y sus consecuencias. Una parte de esas informaciones describen datos experimentales contrastables, como el aumento en la concentración de CO2 en la atmósfera, el aumento de la temperatura global del planeta o la disminución del pH en los océanos. Pero una parte no menos importante de esas informaciones se refieren a predicciones sobre lo que va a pasar con esas cuestiones en un futuro más o menos próximo que, por el momento, suele cifrarse con fecha tope en 2100. Se supone que van destinadas a nuestros políticos, para que tengan argumentos a la hora de tomar medidas paliativas ante los efectos de esos cambios, pero creo que a nadie se le escapa que el tratamiento que los medios hacen de esos pronósticos es, muchas veces, bastante catastrofista.
Las mencionadas predicciones se realizan a base de modelos matemáticos, más o menos sofisticados, a los que viene bien disponer de datos experimentales del pasado para comprobar su fiabilidad. Solo así puede uno tener una idea razonable de las posibilidades de ese modelo a la hora de ponerlo a trabajar en la predicción del futuro, algo siempre complicado y, especialmente, en un sistema tan caótico como el clima a largo plazo.
El problema es que no tenemos tantos datos sobre el pasado. Por ejemplo, la magnitud emblemática en el calentamiento global, la temperatura en diferentes lugares de nuestro planeta, se empezó a medir a finales del siglo XVII desde cuando, excepcionalmente, tenemos registros de lo que se conoce como Temperaturas de la Inglaterra Central (CET). En otros países existen también estaciones emblemáticas que acumulan datos desde mediados del XIX o posteriores. El observatorio de Igueldo, en mi pueblo, comenzó en fecha relativamente tardía (1928) aunque es de los más veteranos de España. Pero muchas de esas estaciones tempraneras pertenecen al Hemisferio Norte, con lo que deducir de ahí una temperatura global de la Tierra y su evolución durante los pasados decenios o siglos es realmente aventurado. Y ya no digamos de registros de la temperatura de los océanos, principales constituyentes del planeta azul, de los que casi no ha habido registros hasta muy recientemente. Y lo mismo pasa con otras medidas que ahora nos interesan, como las mencionadas relativas a la concentración de CO2, al pH de los océanos o a cosas como el nivel del mar.
Pero, como dicen los meteorólogos, el clima no es "el tiempo". Predecir el clima hasta 2100 (o más allá) no es predecir el tiempo meteorológico a cinco días vista. Y los modelos necesitan, como he mencionado antes, tener datos de muchas variables, extendiéndose muchos siglos hacia atrás, para comprobar previamente la fiabilidad de sus predicciones futuras. Y ello solo es posible, indirectamente, gracias a una serie de herramientas que se ha dado esa rama de la Ciencia que llamamos Paleoclimatología y que se agrupan bajo la denominación de Indicadores Climáticos (o Climate Proxies, en la literatura en inglés). Como muchos de esos indicadores están basados en conceptos y técnicas analíticas ligadas a la Química, bien merece que hablemos aquí de ellos. Aunque, me temo, eso va a dar para más de una entrada.
Los testigos de hielo (Ice Cores) tomados de glaciares y regiones del planeta permanentemente cubiertas por hielo, son una excelente fuente de indicadores climáticos con resonancias químicas. Por ejemplo, tanto Groenlandia como la Antártida tienen espesores de hielo de tal magnitud y pureza que nos permiten extraer de ellas cilindros de hielo, como el que veis en la figura que ilustra esta entrada (y que podéis ampliar clicando en ella). Estos gélidos "chorizos" permiten estudiar diversas variables acumuladas en su interior, en intervalos de tiempo que cubren 100.000 años en el caso de Groenlandia y 400.000 o más en el caso de la Antártida.
Con esos "testigos" en el laboratorio son varias las variables ligadas al calentamiento global que podemos estudiar. Por ejemplo, la composición de la atmósfera y su evolución. Cuando en estos lugares de climas extremos nieva, nuevas capas de hielo se generan a partir de la nieve y se acumulan sobre las anteriores. En el tránsito entre los copos de nieve y el hielo consistente, el aire de la atmósfera del momento queda ocluido en el hielo y, con él, los diferentes gases constitutivos de esa atmósfera, nitrógeno, oxígeno, CO2 y, en menor medida, otros gases como el metano y óxidos de nitrógeno.
Así que si capturamos por algún procedimiento esos gases a diferentes niveles de los "chorizos" de hielo y los analizamos, podemos reconstruir la evolución de la composición de la atmósfera en esos gases a lo largo del tiempo. Y así si vais a este enlace, podéis ver la evolución en la atmósfera en lo relativo al CO2,, metano y óxido de nitrógeno en uno de los testigos más mencionados en la literatura, obtenido en la Antártida y que cubre los últimos 800.000 años. Y si ya os centráis, a partir de esos datos, en el último milenio, parece evidente que, sobre todo en las últimas décadas, el contenido de la atmósfera en esos gases ha crecido de forma clara, particularmente en lo relativo al CO2, cuya concentración está expresada en partes por millón (ppm), mil veces superiores a las partes por billón (ppb) de los dos otros gases. Ese crecimiento ha sido confirmado en años recientes (a partir de 1959) por las medidas realizadas en el Laboratorio hawaiano de Mauna Loa, dedicado a este fin.
Pero el hielo de los testigos proporciona mucha más información. El hielo es agua y, en tanto que agua, sus moléculas están constituidas por átomos de hidrógeno y átomos de oxígeno. Aquí, para los que ya han olvidado su Química del bachillerato, tengo que recordar que cualquier átomo está constituidos por protones, electrones y neutrones. Estos últimos, los neutrones, para un determinado átomo, pueden variar, dando lugar a los llamados isótopos. Por ejemplo el hidrógeno más común tiene un protón, un electrón y ningún neutrón, pero hay un isótopo del hidrógeno que tiene un neutrón en su nucleo y que llamamos Deuterio. Lo mismo pasa en el oxígeno. El oxígeno normal (oxígeno-16) tiene 8 protones, 8 electrones y 8 neutrones, pero el llamado oxígeno-18 tiene 10 neutrones y no ocho. Como consecuencia de ello, un átomo de deuterio pesa más que un átomo de hidrógeno normal y un átomo de oxígeno-18 pesa más que uno de oxígeno-16.
La mayoría de las moléculas de agua están constituidas por un oxígeno-16 y dos hidrógenos normales. Pero hay otras combinaciones de isótopos de hidrógeno y oxígeno que dan lugar a "otras aguas". Las más relevantes para esta discusión son la constituida por dos hidrógenos normales y un oxígeno-18 y la constituida por un hidrógeno normal, un deuterio y un oxígeno-16. Estas dos últimas, y muy minoritarias, formas del agua son más pesadas que el agua de toda la vida que llena ríos, lagos y océanos de la Tierra. Cuando el agua de estos lugares se evapora, el agua normal y mayoritaria lo hace con mayor facilidad que las dos formas minoritarias, por ser menos pesada, con lo que el vapor de las nubes que se forman en esos lugares suele llevar una proporción del agua convencional superior a la que se da en los océanos. Pero cuando ese vapor viaja en forma de nubes hasta los polos y condensa para caer en forma de nieve, caen en mayor proporción relativa las moléculas más pesadas de agua, las que contienen los isótopos deuterio y oxígeno-18. Este lío de diferentes moléculas de agua hace que el análisis de la concentración de deuterio y oxígeno-18 en el hielo de los testigos permita reconstruir la velocidad a la que el agua se ha evaporado desde los océanos y ha acabado como nieve en los polos. Como la evaporación del agua y posterior caída en forma de nieve depende de la temperatura, uno puede reconstruir la temperatura ambiente mediante los análisis mencionados. Un poco complicado pero creo que suficientemente entendible.
Este tipo de análisis ha permitido, por ejemplo, reconstruir la temperatura de los últimos 800.000 años a partir de las variaciones en la composición en deuterio de testigos de hielo tomados en la Antártida. Y así, en esta figura, se muestran esos datos junto con la evolución de la composición en CO2 de la atmósfera, obtenida a partir del análisis del aire atrapado en esos mismos testigos. Se ve que hay una correlación entre uno y otro tipo de datos, mostrando los cambios que ambos han sufrido a lo largo de ese extenso período de tiempo.
A raíz de este tipo de gráficas hay algunos encendidos debates sobre qué fue antes "el huevo o la gallina" y las implicaciones que eso tiene en la situación actual. Aunque, en principio, parece que en épocas pasadas la subida de temperaturas fue anterior a la subida en la concentración del CO2, dicen algunos de los que saben de esto que, en el momento presente, esa correlación puede invertirse al ser nosotros los que estamos forzando la subida en la concentración de ese gas.
Pero hasta ahí no llego. El Búho necesitaría otra vida para leer mucho más sobre estas cosas y tener una opinión fundada al respecto. Aunque sobre otros indicadores climáticos creo que lo tengo más claro. Pero eso da para otro post.

Dadas las proclamas que envuelven el marketing de los perfumes, seguro que muchos de vosotros pensáis que la mayoría de los productos de marcas caras y conocidas son un delicado equilibrio entre extractos de diferentes flores, conseguido gracias a ese portentoso órgano que llamamos nariz y que es la herramienta más potente de los perfumistas más afamados. Pues bien, eso no es así. Salvo raras excepciones, el adjetivo más representativo para denominar a esa compleja mezcla de aromas que llamamos perfume podría ser el de semisintética. Es decir, algunos de sus componentes se extraen, efectivamente, de flores y plantas pero otros muchos son de carácter sintético, comercializados por grandes empresas dedicadas a ello. Y que no sólo han generado moléculas nuevas con aromas característicos e innovadores, sino que han reproducido en el laboratorio míticas moléculas de determinadas esencias, obtenidas a partir del mundo vegetal (o animal, pero eso es otra historia que conté hace poco). La proporción entre uno y otro tipo de componentes puede variar pero, a nivel global, podemos decir que casi el 90% en peso de lo que se emplea en un perfume es de origen sintético. Como estos productos son más baratos que los extraídos de plantas muy codiciadas por los perfumistas, muchas veces el precio es indicativo de la cantidad de extractos naturales que llevan.
Y sobre una molécula, originariamente extraída de la naturaleza y hoy usada, en su mayor parte, como sustancia química pura producida a nivel industrial, va esta entrada. Se trata de la cumarina, una molécula clave en el desarrollo de la industria del perfume a finales del siglo XIX. Pero antes, vamos a enfocar la entrada desde otro punto de vista, lo que estoy seguro placerá mucho a mis lectores de Irún, donde tengo algún que otro fiel seguidor.
Pravia, como muchos de vosotros seguro que sabéis, es una localidad asturiana. Pero probablemente no la asociéis a la marca comercial Heno de Pravia, ese conjunto de productos de perfumería desarrollados por la empresa creada por un irundarra de pro, Salvador Echeandía Gal, bajo el nombre de Perfumería Gal, que después fue englobada en el grupo Puig. A los futboleros os sonará el nombre del Stadium Gal, donde juega el Real Unión de Irún, en unos terrenos cedidos por el propio Echeandía. Dice la historia que en 1903 el mencionado Echeandía, junto con un amigo, Lesmes Sainz de Vicuña, también originario de la villa fronteriza, andaban de viaje por Asturias cuando quedaron prendados del olor a hierba recién cortada en un prado cercano a Pravia. Y no pararon hasta reproducir ese aroma en un producto, su jabón Heno de Pravia, que aún hoy en día se vende como rosquillas y prácticamente en todo el mundo, junto a otros productos como colonías, desodorantes, cremas de afeitado. Y la Química tuvo mucho que ver en este asunto, entre otras cosas porque otro Echeandía Gal, Eusebio, era un químico muy viajado para la época, con estudios en Berlín.
El día de fin de año de 2013, os contaba yo el descubrimiento por chiripa de la mauveína, un producto que abrió la veda a una gran parte de la gama de colorantes sintéticos que hoy se emplean en el mundo. El autor del descubrimiento fue un imberbe (18 añitos) William H. Perkin, que andaba a la búsqueda de la quinina y que por chiripa y a espaldas de su jefe, A.W. von Hoffmann, llegó a la mauveína. Mantuvo en secreto el descubrimiento, lo patentó y creó un imperio que le hizo lo suficientemente rico a los 30 como para dejar sus negocios a la familia y seguir dedicándose a lo que más le gustaba, generar nuevas moléculas de potenciales aplicaciones.
Y como no tenía un pelo de tonto, enseguida se dio cuenta de que uno de los productos que obtuvo en esos primeros años de andar a la suyo (1868), fue la cumarina, una sustancia que había sido aislada en los años veinte del siglo XIX a partir de la llamada Haba Tonka, que no es otra cosa que la semilla de un árbol llamado Dypterix odorata. Como en los casos de la vainilla y de la goma garrofín, que ya hemos visto en otras entradas, son esas vainas secas las que acumulan diversas sustancias que constituyen su aroma característico. En el caso que nos ocupa y entre otras sustancias constitutivas, el principal aroma del Haba Tonka lo da la cumarina, que se encuentra en ella en un porcentaje entre el 1 y el 3%. En cualquier caso, no son esas habas la única fuente de este aroma, igualmente presente en cosas como la vainilla, en la canela, en algunas variedades de hierba, en los tréboles, etc.
La irrupción de la cumarina sintética de Perkin en el mundo de la perfumería se produjo en 1882 en el famoso y hoy ya desaparecido Fougère Royal, una creación del perfumista Paul Parquet para la empresa Houbigant. Parquet usó cumarina en una concentración al 10%, junto con lavanda, geranio y musgo de roble en un intento de reproducir el aroma de los helechos (Fougère en francés). Desde entonces, han sido casi innumerables los perfumes que han seguido esa senda, perfumes que suelen agruparse dentro de la gama Fougères. Por ejemplo, la cumarina ha sido y sigue siendo un componente fundamental en la paleta olfativa de la casa Guerlain, en perfumes como el mítico Jicky o su famoso y selecto Tonka Imperiale, pero otras muchas casas famosas lo han empleado (Dior, Chanel, Givenchy...). Aunque no está del todo claro en la historia de la génesis del Heno de Pravia, muchos que saben del tema piensan que algo oyó Echeandía en la época sobre las ideas de Parquet y con ayuda del químico de la familia lo aplicó a su búsqueda del jabón que le ha hecho famoso. El nombre cumarina sigue apareciendo en algunas etiquetas de la marca Heno de Pravia, aunque por razones que explico debajo, quizás se hayan buscando fórmulas alternativas a su uso.
Porque si hoy Parquet se hubiera puesto a crear el Fougère Real no lo hubiera tenido fácil. Como otros muchos componentes de las cosas que nos aplicamos para oler bien, la cumarina ha tenido sus problemas "sanitarios" ligados a la creciente quimiofobia que nos invade y que hace que cualquier sustancia sintética sea mal vista. Es verdad que durante años estuvo envuelta en una polémica ligada a su carácter a su carácter hepatotóxico en ratas. O a la llamada "enfermedad del trébol dulce" que acababa con los conejos a base de hemorragias. Hoy sabemos que, bajo la acción de ciertos mohos, la cumarina contenida en los tréboles genera dicumerol, un potente anticoagulante. Sabemos también que los problemas de las ratas con la cumarina son mucho más graves que los que causa en los humanos, en los que es moderadamente tóxico para el hígado y el riñón, pero tendríamos que comer muchas habas de Tonka o cumarina pura todos los días para que la cosa fuera grave. También, como en otros muchos componentes de los perfumes y colonias, algunos estudios achacan a la cumarina un carácter alérgeno, por lo que la IFRA (International Fragance Association) restringe su uso en perfumes (categoría 4) a una concentración del 1,6%. Pero, digámoslo una vez más, esa restricción se aplica a la cumarina venga de las habas Tonka o de la cumarina pura y sintética vendida por las industrias que comercializan aromas.
Y esta disquisición final me lleva a comentar algo que me ha sorprendido últimamente en mis búsquedas en la red de material sobre la Química de los perfumes. En los foros de muchas webs dedicadas a estos temas, he descubierto que la gente se compra aceites esenciales y se prepara sus propios perfumes. No me parece mal como entretenimiento y búsqueda de aromas personalizados. Pero, ¿se mirarán las normas de la IFRA en lo relativo a cantidades máximas seguras a emplear de cada uno de esos aceites esenciales?.

Mi ya finiquitada actividad investigadora ha estado centrada, durante casi cuarenta años, en el mundo de los polímeros, unos materiales que mucha gente simplifica en plásticos, algo inadecuado pero que no toca explicar ahora. Por ello, no es de extrañar que todavía me siga interesando todo lo que se cuece en torno a estos materiales que eclosionaron a lo largo del siglo XX. La mayor preocupación actual sobre ellos es qué hacer con sus residuos y, entre los varios frentes de esta problemática, está el tema de los residuos de plástico que se vierten al mar. Durante su vida en el mismo, muchos plásticos se fragmentan en trozos pequeños que se conocen, genéricamente, como microplásticos y que, en años recientes, han sido objeto de diversos estudios que muestran su presencia incluso en recónditos sitios de nuestros océanos y su impacto sobre la vida animal de los mismos.
Llevo semanas estudiando un voluminoso libro de casi 500 páginas titulado Marine Anthropogenic Litter (Basura Marina de Origen Humano, para que nos entendamos) una obra colectiva editada por Melanie Bergmann, Lars Gutow y Michael Klages y publicada en 2015 por Springer International Publishing. No me resulta fácil, porque en algunos de los temas estoy un poco pez, pero ando tomando interesantes notas de cara a ponerme al día y a hacerme una idea global del asunto para luego contar algo aquí. Pero, como aperitivo, os voy a contar algunas ideas al hilo de una noticia que me llegó por un tuit de ese monstruo de la divulgación científica que es Paco Villatoro (@emulenews). El tuit hace referencia a un artículo publicado online este 17 de agosto en los Scientific Reports de Nature por investigadores del Departamento de Ingeniería Química de la Universidad de Alicante. El artículo resume un estudio de los microplásticos presentes en diversas sales de mesa comercializadas en España. Un estudio similar ya se había hecho hace dos años sobre las sales comercializadas en China y otro se acaba de publicar en un numero precedente (en abril) del mismo Scientific Reports. Como consecuencia de ello, internet contiene ya algunos ejemplos de proclamas sensacionalistas sobre estos estudios cuando, creo yo, no debiera ser el caso.
Voy a comentar el artículo de los colegas alicantinos porque contiene algunos resultados que me han sorprendido. Se han estudiado 21 sales comercializadas en España, fundamentalmente provenientes de salinas marinas tanto del Atlántico (no dan marcas pero si localizaciones en Andalucía, Canarias y Galicia) como del Mediterráneo (Cataluña, Valencia, Murcia, Alicante, Baleares), así como tres sales de manantiales salinos en el interior, alejados del mar. Uno me cae cerca (Añana, en Alava, de cuya sal ya hablamos hace años aquí), los otros dos están localizadas en las provincias de Cuenca y Alicante. Esta última puede ser de la zona de Villena, una zona salinera con tradición centenaria. En el estudio, esas muestras se disuelven, se centrifugan para eliminar arena y otras partículas sólidas más densas, se filtran con filtros que no dejan pasar partículas de tamaño superior a 5 micras y los microplásticos así recogidos se cuantifican con ayuda de microscopios y se identifica su naturaleza mediante técnicas de análisis de plásticos convencionales (FTIR).
El primer resultado que me ha sorprendido es que tanto la sal de mar como la proveniente de salinas interiores contienen cantidades similares de partículas de microplásticos, (las de mar entre 50 y 280 micropartículas por kilo de sal y las de interior entre 115 y 185). Como me conozco bien la estructura del Valle de Añana y cómo se generaron en su día los manantiales que proporcionan agua con altos contenidos en sal (salmuera), el resultado es como para pensárselo.
Debéis saber que el Valle de Añana, hace más de 200 millones de años, estaba bajo el mar. La evaporación posterior del agua en ese valle cerrado generó una gran cantidad de sal que posteriormente fue cubierta por otros estratos. La posibilidad actual de obtener sal en Añana se explica por el fenómeno geológico denominado diapiro. En líneas generales, consiste en la ascensión hacia la superficie terrestre de materiales más antiguos debido a su menor densidad. El diapiro de Añana atraviesa en su ascenso algunos de los acuíferos más importantes de la región, por lo que se convierten en puntos de salida de las aguas confinadas en los mismos. Uno de los puntos de salida es el manantial de Santa Engracia, donde el agua es una muera que sale en carga, es decir a mayor presión que la de la superficie. Esto explica la gran saturación en sales, disueltas en los lechos salinos atravesados durante el ascenso del agua a lo largo de la chimenea diapírica, en condiciones de presión y temperatura superiores a las de superficie. Este manantial es el punto de partida de la explotación de Añana, con una salinidad media superior a 250 gramos por litro. Y no deja de sorprenderme que, si el proceso es así y está recogido en sesudos trabajos geológicos, ¿de dónde provienen los microplásticos de la sal de Añana?. Porque hay mucho "filtro" en todo el proceso. El artículo no hace reflexión alguna al respecto. Así que si alguien tiene constancia de las razones, no vendría mal que nos lo explicara en los comentarios.
Otro resultado que me llamó inicialmente la atención es la composición de los microplásticos analizados en la totalidad de las sales consideradas. El 83% es polietilen tereftalato (PET), el plástico empleado en las botellas de agua, bebidas de cola, etc., mientras que el resto corresponden, fundamentalmente, a microplásticos de polietileno y polipropileno, mucho más vendidos que el PET. En las de la sal china arriba mencionada, el componente más abundante es el celofán (que allí se sigue utilizando como envase), mientras que las cantidades de PET, PE y PP son más concordantes con su importancia en el mercado. Dicen los autores, y no me parece mal, que esto pudiera deberse a que la mayor densidad del PET le hace permanecer en el agua desde la que está cristalizando la sal durante todo el proceso, mientras que PE y PP, de menor densidad y que deben flotar en la salmuera, podrían ser arrastrados por los operarios durante las labores de manejo de lesta y ser, por tanto, eliminados más fácilmente de la misma.
El artículo termina con un apartado en el que los autores tratan de evaluar el impacto en nuestra salud de los microplásticos contenidos en la sal que consumimos, que es lo que imagino que a la mayoría de vosotros le interesa más, a la vista del título que le he puesto a la entrada. Sobre la base de un sencillo cálculo ligado al consumo de sal recomendado por la Organización Mundial de la Salud, los autores establecen que podemos meternos al coleto del orden de 500 piezas de microplástico por año, lo que les parece una cantidad irrelevante. Y estoy de acuerdo, porque, en primer lugar, esos trocitos de plástico no los digerimos y por tanto saldrán tal cuales, igual que otras cosas indeseadas que digerimos, como, por ejemplo, los sólidos contenidos en chipirones o chopitos poco limpios, almejas y similares.
En ese mismo apartado, el artículo hace referencia a los dos temas que siempre aparecen en los artículos relacionados con la contaminación de plásticos en el mar. El primero, los problemas que puede causar a pájaros y peces la ingestión de los mismos. Dependiendo del tamaño del ser vivo y del trozo de plástico engullido, el asunto puede llegar a la muerte del animal, pero no por contaminación sino por simple bloqueo de su tracto gastrointestinal.Y, de nuevo, no solo plásticos ingieren pájaros y peces creyendo que les pueden alimentar.
El otro asunto es más sutil y se basa en que los microplásticos pueden actuar como "refugio" de sustancias tóxicas con carácter lipofílico (aquellas que se disuelven mejor en grasa que en agua). Y entre ellas se suelen citar, sobre todo, a los Contaminantes Orgánicos Persistentes (POPs en inglés) como los bifenilos policlorados o las dioxinas y sus parientes los furanos. Cuando esos microplásticos y su carga de POPs son ingeridos por animales, la teoría es que estas sustancias se introducen en la cadena de alimentación y llegan hasta los humanos.
Yo soy bastante escéptico sobre el asunto (al menos cuantitativamente), pero necesito ordenar mis ideas y en ello estoy con el libro arriba mencionado. Si llego a alguna conclusión, sea políticamente correcta o no, la contaré. Mientras tanto, no me sean guarros y lleven los plásticos que ya no puedan usar al correspondiente contenedor. Y si son padres o abuelos, ilustren a su descendencia en este problema. Yo lo sufro todos los días en mi portal con una tienda de chuches adyacente.

Todo comenzó hace ya algunos meses (bastantes). Mi comadrona estaba viendo uno de esos programas de la tele que le gustan, donde se muestran las casas más fabulosas del mundo mundial. En este caso se trataba de una casa en Marbella, propiedad de uno de esos rusos enriquecidos por el petróleo. Una casa que, por tener, tenía hasta un helipuerto y dos hoyos de golf. Aparte de envidias no explícitamente declaradas y que mis lectores intuyen, lo que captó mi atención es que, al enseñar su casa, la rusa del magnate mostró, entre otras cosas, un aparato adosado al grifo de agua de su cocina, con pinta de dispositivo electrónico de calidad y que, según explicó, dividía el agua de grifo en dos tipos de aguas: una alcalina, buenísima para la salud y otra ácida que viene bien para fregar, limpiarse el cutis, etc... Me quedé con la idea de enterarme sobre el asunto pero la cosa, como otras muchas, no ha pasado hasta ahora de unas notas amontonadas en mi escritorio. Hasta que, desde hace unas semanas, se me ha pedido hasta tres veces mi opinión sobre temas relacionados con estos dispositivos.
Cuando me puse a investigar en serio, resulta que estos mal llamados ionizadores de agua tienen una larga historia que se remonta a principios de los años cincuenta, aunque ha sido en las últimas décadas cuando la cosa ha explotado en el mercado, de la mano de diferentes empresas y científicos, fundamentalmente japoneses. Basta con que escribáis water ionizers en Google, seleccionéis el apartado imágenes y os aparecerán cientos de dispositivos existentes en el mercado. Los precios pueden variar de marca a marca, pero los más reputados (no voy a dar nombres para que no se metan conmigo) pueden llegar a costar casi 6000 €. En un artículo de 2013 que tengo encima de la mesa, se cifra en unos 200.000 el número de estos dispositivos que se venden al año y muchos de ellos, sobre todo en USA, se venden por el sistema de hacer reuniones con potenciales clientes a los que convencer de las bondades del aparato y endosarles uno (tal como se hizo en su día con Tupperware).
He puesto al inicio de esta entrada una imagen genérica de cómo funcionan estos aparatos (podéis ampliarla clicando en ella). Básicamente toman agua de grifo, la filtran en un filtro clásico de carbón activo, una forma de eliminar el cloro presente, que podría dañar los electrodos de la celda electrolítica, corazón del aparato y de la que ahora hablaremos. Después del filtrado y si el agua no contiene de origen una cantidad sustancial de sales disueltas que aseguren la conductividad de la misma, se regula ese contenido mediante una serie productos que los suministradores del aparato también venden. Finalmente, ese agua así acondicionada, se mete en la llamada cámara o celda electrolítica, donde se le somete a un proceso de electrolisis, que la descompone en hidrógeno y oxígeno.
No voy a dar muchos detalles técnicos, pero a los iniciados les diré que el sistema electrolítico consta de los clásicos ánodo y cátodo, generalmente de titanio recubierto de platino. Es importante reseñar que esos electrodos se encuentran en compartimentos separados por un diafragma semipermeable hecho de plástico. Ello permite que en el ánodo se genere oxígeno (O2) gas, iones hidrógeno positivos y se acumulen aniones del tipo bicarbonato, cloruro, nitrato, etc, dependiendo de las sales que contenga el agua de partida. En el cátodo se genera hidrógeno gas (H2), iones hidroxilo y cationes tipo sodio, potasio, calcio o magnesio, provenientes igualmente de las sales disueltas. Eso hace que del compartimento anódico salga agua más ácida que la de partida y con un cierto poder oxidante, mientras del cátodo salga agua alcalina de poder reductor o, como suele denominarse en la propaganda de estos aparatos, antioxidante.
En todo lo anterior, este vuestro Búho tiene poco que objetar. El escepticismo comienza cuando, en el marketing de las casas vendedoras, empezamos a leer las maravillas ligadas al empleo de estos ionizadores. En primer lugar, las derivadas de beber agua alcalina (con pH superior a 7), una más de las opciones a la hora de seguir la llamada dieta alcalina, que os estarán vendiendo por todos los lados, ya sea en la tele, en la radio, en los magazines de fin de semana o, sobre todo y como siempre, en internet. El pH de los diversos tipos de agua (de grifo, manantiales naturales, aguas de mesa,..) puede variar bastante y así lo reconoce la legislación española en el Real Decreto 140/2003 Anexo I, que regula el pH de las aguas destinadas a consumo humano. En el mercado podéis encontrar aguas ácidas como la de Vichy Catalán (pH=5,9) o básicas (alcalinas) como la oscense Agua Veri (pH=9,5). Pero todo esto que cuentan de la dieta alcalina es una patraña más de las muchas que giran en torno a nuestra alimentación. Comer o beber cosas alcalinas no altera el pH general de nuestro cuerpo, que tiene un mecanismo primario para controlarlo mediante la exhalación de CO2, que gobierna la cantidad de ácido carbónico en la sangre y, a partir de ahí, de una serie de equilibrios químicos bien establecidos.
Pero el marketing perverso de los fabricantes de ionizadores va un poco más lejos. Debéis saber que el agua alcalina de su ionizador no es agua alcalina cualquiera (es decir con pH superior a 7), como las de algunas de fuentes naturales que ya salen alcalinas de origen (y que acabo de mencionar) u otras a las que se adiciona un exceso de bicarbonato para aumentar su pH. La producida por el ionizador (que ellos llaman agua alcalina ionizada) tiene sus peculiares propiedades debido a que, además del alto pH, hay otros "cambios" generados en ella como consecuencia del proceso que ocurre en el interior del aparato. Y ello, proclaman, hace que sea efectiva en la eliminación de antioxidantes, en tratamientos de cáncer, en la prevención y curación de la diabetes, Parkinson, arterioesclerosis, problemas degenerativos de la retina y otros (ver, por ejemplo, aquí, donde se cantan todas esas bondades, sin soporte científico alguno). Las razones aducidas tienen un doble origen que vamos a desmenuzar aquí.
Como probablemente sepáis, el agua es una molécula polar, lo que quiere decir que tiene partes de ella con cargas ligeramente positivas (localizadas en sus dos hidrógenos) y negativas (el oxígeno), como podéis ver aquí. Ello hace que las partes del agua con distinta carga de diferentes moléculas puedan atraerse y permanecer cerca, formando los llamados agregados o clusters. Su vida es efímera, porque esos agregados se están juntando y soltando continuamente, en periodos de tiempo tan pequeños como las billonésimas de segundo. Todo esto está muy estudiado, desde hace tiempo, por la Química Física y se sabe que esos agregados suelen implicar a unas 12-14 moléculas de agua al mismo tiempo. Los que me hayáis seguido en las entradas sobre la homeopatía recordaréis que la famosa "memoria del agua" se basa precisamente en esos agregados, capaces de retener la forma de la molécula del "principio activo" una vez que ha desaparecido por dilución y, en la lógica homeopática, capaces de curar igual que esa molécula que ya no está.
Pues bien, muchos vendedores de ionizadores proclaman que su aparato es capaz de disminuir ese tamaño habitual de los agregados de agua y dejarlo en 4/6 moléculas. Según ellos, y debido a ese menor tamaño, el agua penetra con mayor facilidad en los diversos componentes de las células y permite una hidratación mejor del organismo. Como otros muchos bulos sobre el posible efecto de fuerzas eléctricas o magnéticas que alteran la estructura conocida del agua, vuestro Búho no se cree esta patraña de los clusters pequeñitos, por mucho que para vestirlo, en alguna de las webs que lo proclaman, aparezcan prestigiosos médicos como Hiromi Shinya, un experto colonoscopista, autor del muy vendido libro La Enzima Prodigiosa, libro e ideas que provocaron que mi amigo JM Mulet (todo un Petronio) fuera tildado de gordo por la inefable Mercedes Milá.
Este asunto de los clusters de menor tamaño ya es un poco viejo. En una de mis contribuciones a Naukas, os contaba el caso de un agua de propiedades maravillosas, vendida en USA bajo el nombre de Penta, que atribuía sus virtudes a clusters más pequeños que los habituales. Tras muchos avatares, que han incluido una demanda ante la Corte Suprema de California, el agua se sigue vendiendo con el mismo nombre pero su marketing se basa, exclusivamente, en la extremada pureza de la misma. En otros casos, y para vender las bondades de los productos de una conocida marca de cosmética, se aducen cambios de estructura en los agregados, esta vez en forma de icosaedros.
La otra pata de los atributos del agua ionizada por estos aparatos es el hidrógeno gas que se produce en el compartimento catódico y que sale acompañando al agua alcalina. En 1985, el Director del japonés Instituto del Agua, adquirió un ionizador y comenzó a estudiar las propiedades del agua por él producida, utilizándola como bebida habitual y preparación de comidas en la Kyowa Medical Clinic. Según él, observó una clara mejoría en pacientes que sufrían trastornos gastrointestinales y otras patologías ligadas al hígado. Para explicarlo, en 1995, propuso la hipótesis de que esas mejoras se producían como consecuencia del gas hidrógeno generado en los ionizadores, que ejercía un activo papel de "cazador" de los radicales libres que nuestro organismo genera como consecuencia del oxígeno que continuamente estamos inspirando.
He investigado poco al respecto pero mis genes escépticos han encendido ya ciertas luces de alarma. Así que vamos a dejar que pase un poco de tiempo antes de escribir algo al respecto. Mientras tanto y aunque cada uno se gasta el dinero en lo que quiere, no os recomiendo una inversión en uno de esos ionizadores. Es cierto que tienen una pinta muy de diseño tecnológico y, sin duda, dará a vuestra cocina un tono de lo más californiano. Pero dudo que vuestra salud note algo.

Los que tenemos ya muchos calendarios podemos valorar mejor que los más jóvenes lo que supone disponer de una máquina tan discreta y eficaz como un congelador. Tener guardados alimentos que se conservan más tiempo y que pueden sacarse en un momento, programado o de necesidad, es algo que cambia, literalmente, nuestro modo de comer. Aunque algunas veces se nos presenten algunos problemillas, como el de tener una urgencia para utilizar algo que teníamos congelado y no tener tiempo para descongelarlo mediante un proceso lento y seguro. Esa "molesta" situación es la que me planteaba un amigo hace unos días, pidiéndome alternativas.
Este Búho ha usado mucho el congelador, porque para la ajetreada vida que hemos llevado cuando éramos más jóvenes, comer todos los días en casa era difícil de conseguir si no lo usabas racionalmente. Y en esa necesidad, me he encontrado bastantes veces en la situación de mi amigo. Pronto aprendí que la descongelación mediante un microondas no era una solución satisfactoria porque, en muchas ocasiones, siempre acababan por empezar a cocerse las partes más delgadas del alimento a descongelar.
Pero como fiel seguidor de mi amigo Harold McGee, hace ya seis años pude leerle en el New York Times sobre los experimentos que había llevado a cabo para descongelar viandas en tiempos más o menos razonables y que resultan adecuados cuando se te ha olvidado dejarlas la noche anterior en tu frigorífico, la forma más aconsejada de descongelar. Y el truco es sencillo: consiste en introducir tu alimento congelado en una de esas bolsas herméticas de plástico, las que llevan una cremallera o similar, y colocar esa bolsa y su contenido en un recipiente con agua no excesivamente caliente. En cuestión de minutos tu alimento estará descongelado. Supongo que muchos de vosotros conoceréis esa posibilidad pero vamos a documentar un poco las razones de la misma.
La culpa de todo la tiene Joseph Fourier, un matemático y físico francés con una curiosa historia y que nos ilustró sobre los misterios de la transmisión del calor. De origen humilde acabó en un orfanato, pero sus educadas maneras y su talento para las matemáticas hicieron que el mismísimo obispo de Auxerre lo colocara bajo la tutela de unos monjes benedictinos, lo que le permitió seguir estudiando. Subyugado por las ideas de la Revolución Francesa se adhirió a ella pero, como muchos de sus correligionarios, cayó finalmente en desgracia y estuvo a punto de poner el cuello debajo de Madame Guillotine. Pasado el susto y las urgencias revolucionarias, sus conocimientos le permitieron acompañar a Napoleón en su campaña en Egipto en 1798. La carrera académica de Fourier estuvo muy ligada al período napoleónico y cuando este se fue a pique, Fourier duró poco.
Pero, para entonces, ya había concebido (1812) una de las obras fundamentales de la Física de la época, la Teoría Analítica del Calor (publicada en 1822), en la que puso en forma de ecuaciones matemáticas el clásico experimento de Jean-Baptiste Biot, en 1804, en el que una barra de hierro se calienta en uno de los extremos y se enfría en el otro, permitiendo que la misma alcance una situación estacionaria, en la que el calor suministrado por el extremo caliente va "fluyendo" hacia el otro extremo a una velocidad que depende del punto de la barra que consideremos. Lo de fluir y otras cosas daría para varias entradas, pero no es el momento.
Lo que aquí interesa es que, de acuerdo con la ley de Fourier de la transmisión del calor, cada material transmite el calor de manera diferente, aún en la situación en la que la diferencia de temperatura entre las partes frías y calientes del mismo sea la misma. Por ejemplo, y para lo aquí nos ocupa, los gases transmiten el calor peor que los líquidos y estos peor que los sólidos. Sobre esto ya os he contado algo en el Blog, cuando os explicaba cómo enfriar champán con una mezcla de agua, sal y hielo, más rápidamente que en un congelador, siempre que se usen composiciones adecuadas de la llamada mezcla frigorífica. Pues aquí es algo parecido pero el efecto inverso.
Cuando dejamos un trozo de salmón o unas pechugas de pollo a descongelar en una cocina que está, como hoy la de mi casa, a 23 ºC, el calor se transmite desde el aire que constituye la atmósfera de mi cocina (una mezcla de oxígeno y nitrógeno, fundamentalmente) a la pieza congelada. Ese proceso de transmisión viene marcado por la llamada conductividad térmica del aire que es, aproximadamente, de 0,02 W/(m-K). Sin embargo, cuando yo pongo mi bolsa de plástico cerrada con mi alimento congelado en un recipiente con agua a la misma temperatura de 23 ºC, la magnitud que controla ahora el mismo proceso de descongelación es la conductividad térmica del agua, más de veinte veces superior a la del aire. Así que, más o menos, esa es la diferencia en los tiempos necesarios para la descongelación.
Si subimos la temperatura del agua, el proceso se acelerará, tanto porque la conductividad del agua aumenta con la temperatura como porque el calor fluye más rápidamente cuando mayor es la diferencia de temperatura entre el cuerpo caliente (agua) y el cuerpo frío (el congelado), así que podríais emplear aguas más o menos calientes para controlar el proceso en tiempos adecuados a vuestras necesidades. Pero no abuséis. Temperaturas más altas implican, en principio, una mayor posibilidad de desarrollo de microorganismos en la superficie de vuestros congelados.
Harold McGee cita en su artículo estudios científicos de la USDA americana [Journal of Food Science 76, 156 (2011)] sobre los tiempos de descongelación de chuletas de buey a diferentes temperaturas del agua, así como las consecuencias que ese proceso tiene en la habitual pérdida de agua del alimento durante el mismo o en otras propiedades ligadas a la textura final de la chuleta. Se hace eco igualmente de estudios de la Universidad de Utah [Food Control 20, 706 (2009)], esta vez sobre pechugas de pollo, en los que además de las características anteriores se fijan en el crecimiento bacteriano y en propiedades organolépticas de pechugas descongeladas frente a otras que no sufrieron el proceso de congelación. En lo que se refiere a lo primero, en piezas relativamente pequeñas como las pechugas, parece que el crecimiento bacteriano quedaba dentro de límites seguros. Y en lo que se refiere al producto descongelado y posteriormente cocinado, parece que un panel de cata no fue capaz de discernir qué pollo había estado congelado y cuál no.
Como consecuencia de todo ello y de sus experiencias, Harold recomienda utilizar una temperatura para el agua en torno a unos 40-50 ºC lo que, generalmente, podéis conseguir jugando con vuestros grifos de agua caliente y fría. Harold recomienda agitar de vez en cuando el agua y comprobar periódicamente cómo va la descongelación, para no exponer la pieza más tiempo del necesario.
La próxima vez que estéis en un apuro lo probáis.











{kind=link}
{kind=link}
{kind=link}
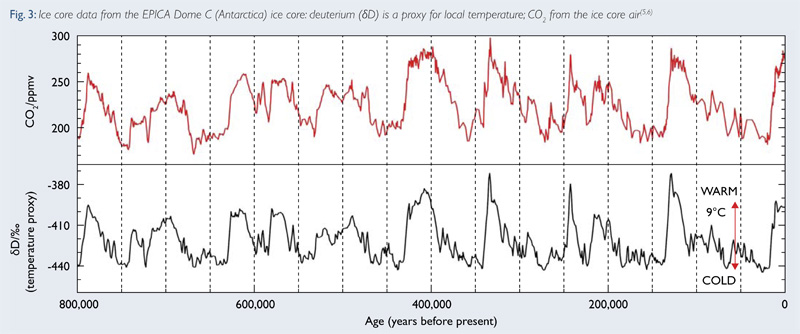{kind=link}
{kind=link}